
RNA-seq受託解析
RNA-seq解析とは
次世代シーケンサー(NGS)を用いて全転写産物の塩基配列を決定することにより、転写産物(RNA)の種類と量を網羅的に検出します。群間で発現変化する遺伝子群の検出や、その遺伝子群が関与する生命現象やシグナルパスウェイを推定することが可能です。
選択したプローブの範囲内でしか変化を検出できないマイクロアレイ法に対して、転写産物を直接シーケンスして発現プロファイルを取得するため、全く予想しなかった発現変化も検出できます。アレイ法に比べて高発現でも飽和しにくく感度も高いため、ダイナミックレンジが広く高い定量性を持っているのも特徴です。
しかしながら、得られた発現量はサンプル(組織や細胞集団)中の平均的な発現量であるため、大多数の細胞の発現情報に埋もれて検出が困難な希少細胞における発現変化を検出できない点には注意が必要です。細胞ごとの発現変化の検出には、シングルセルRNA-seqを始めとしたシングルセル遺伝子発現解析法が用いられます。
用途
- 網羅的な遺伝子発現量推定
- 群間における発現比較(発現変動遺伝子の検出)
- 発現変動遺伝子(DEG)の機能解析(GO解析*1、GSEA*2、パスウェイ解析など)
※関連する解析サービス
シングルセルRNA-seq解析 >
ホルマリン固定後のシングルセルトランスクリプトーム解析 >
高感度なシングルセル完全長total RNA-seq解析 >
シングルセルレパトア解析+シングルセルRNA-seq解析 >
シングルセル空間トランスクリプトーム解析 >
スポット型空間トランスクリプトーム解析 >
このような方に
- シングルセルRNA-seq解析とバルク(通常の)RNA-seq解析の結果を合わせて解釈したい方
- 参考文献と同じような解析を自身のサンプルで実施したい方
- 自身の研究目的に最適な実験デザインの検討から始めたい方
- 共同研究先の対応スピードや、一方向的な情報科学的解析手法にお悩みの方
- 最適なデータ解析手法や適切な統計手法の選択と実施に不安がある方
当社の特徴
- ウェット実験とデータ解析の両分野の経験豊富な専門技術者が直接対応
- 組織からのRNA抽出作業
- サンプル調製のサポート
- 生物学的意義を踏まえたデータ解析(詳細はこちら)
- 論文投稿、学会発表に使用できる各種グラフの作成まで一貫してサポート
- 統計学の専門用語を極力排除した分かり易い説明
- 解析結果の統計学的および生物学的解釈のサポート
解析例
解析例1 マウス新生児の出生後に見られる肝臓の遺伝子発現変化の検出
胎児期から出生後にかけて肝臓の主要な機能は造血から代謝へと変化します。出生直後と10日後のマウス新生児肝臓のRNA-seqデータを使用し、肝臓の機能変化に伴って起こる遺伝子の発現変化を検出しました。発現に変化が見られた遺伝子(発現変動遺伝子、DEG)を抽出し、発現変動遺伝子の特徴を各手法により可視化しました。さらに、発現変動遺伝子に対するGene Ontology(GO)解析、Gene Set Enrichment Analysis(GSEA)により、出生後に起こる肝臓の機能的変化を推定しました。
実験概要の模式図
実験概要の模式図
出生直後(day0)および出生10日目(day10)のマウス新生児肝臓のRNA-seqデータを用いて、マウス新生児の肝臓における遺伝子発現変化を検出しました。
各検体の遺伝子発現量推定結果および主成分分析(PCA)結果
各検体の遺伝子発現量推定結果および主成分分析(PCA)結果
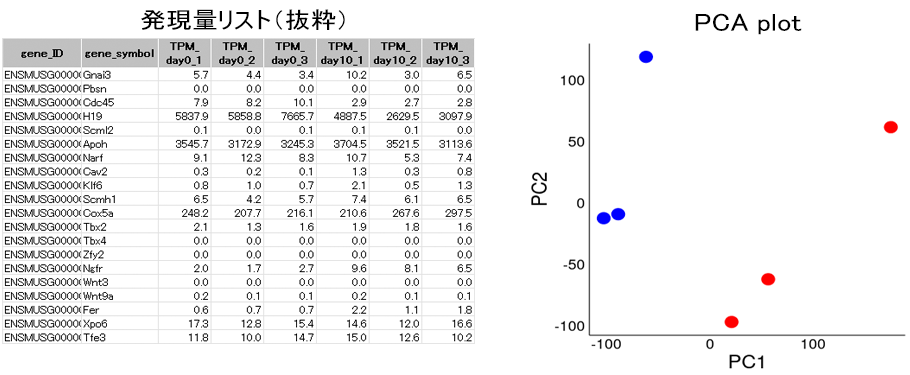
図の左側に示した、各検体の遺伝子発現量リスト(抜粋)をもとに主成分分析を行った結果を右図に示します。青い点はday0、赤い点はday10の各検体を示します。それぞれの群に属する検体には顕著なばらつきは見られませんでした。
2群間の発現変動遺伝子(DEG)リスト (抜粋)
2群間の発現変動遺伝子(DEG)リスト (抜粋)
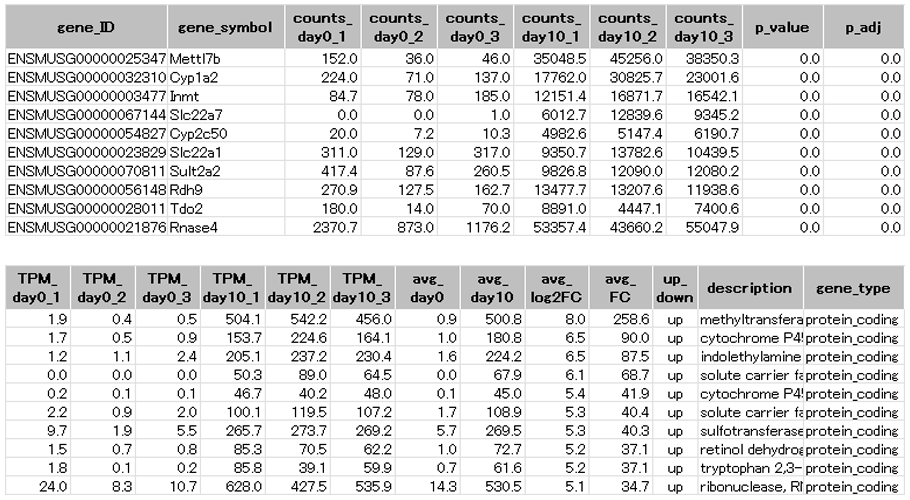
| gene_ID | : | Ensembl遺伝子ID |
| Gene_symbol | : | 遺伝子名の略号 |
| counts | : | 各検体の各転写産物のカウント数 |
| p_value | : | 有意確率 |
| p_adj | : | 補正した有意確率 |
| TPM | : | 各検体の各遺伝子の正規化後の発現量(TPM値) |
| avg_day0 | : | day0群のTPM値の平均 |
| avg_day10 | : | day10群のTPM値の平均 |
| avg_log2FC | : | log2(day0群に対するday10群の発現変動比) |
| avg_FC | : | day0群に対するday10群の発現変動比 |
| up_down | : | day0に対するday10の増減 |
| description | : | 遺伝子のフルネーム |
| gene_type | : | 遺伝子の分類 |

day10で有意に発現量が増加した発現変動遺伝子を抜粋して示しました。
2群間の発現変動遺伝子の可視化(Heatmap)
2群間の発現変動遺伝子の可視化(Heatmap)
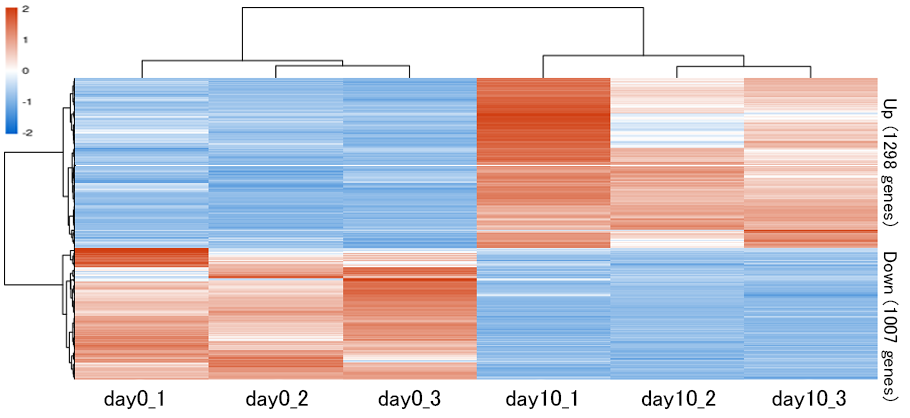
各検体における発現変動遺伝子の発現量をヒートマップで一覧表示しました。各検体の発現量を標準化し、発現量が多い遺伝子を赤色、少ない遺伝子を青色、中間を白色で表示しました。図の縦軸は遺伝子、横軸は検体を表します。
2群間の発現変動遺伝子の可視化(Volcano plot)
2群間の発現変動遺伝子の可視化(Volcano plot)
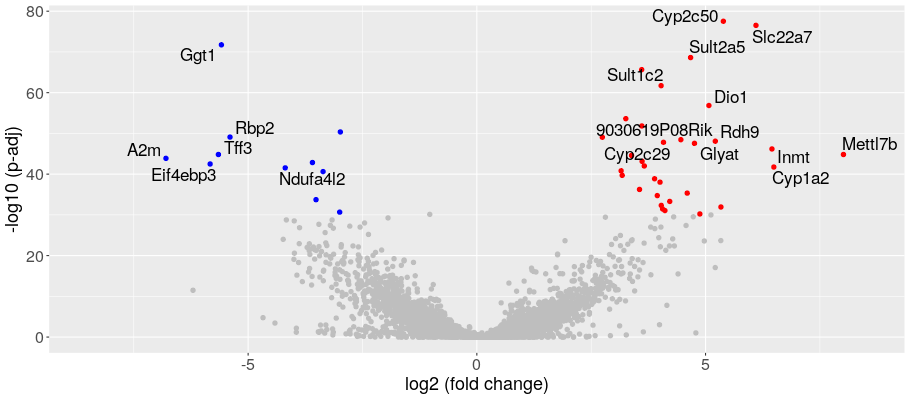
ボルケーノプロットで発現変動遺伝子の発現変動比と統計的有意差を一覧表示しました。縦軸は調整済みp値を-log10で表示し、横軸は発現変動比をlog2で表示しました。各点は遺伝子を表しています。発現変動比と調整済みp値によって分類し色分け表示しました。
2群間の発現変動遺伝子のGO解析
2群間の発現変動遺伝子のGO解析
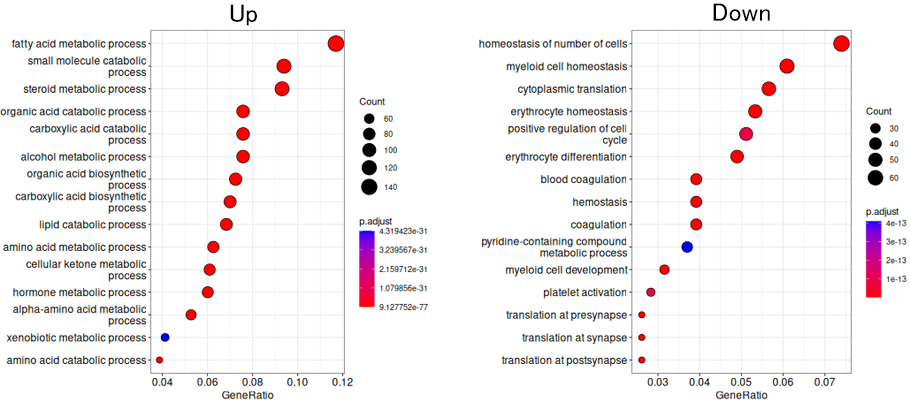
day 10で2倍以上増加(左図)、もしくは減少(右図)した発現変動遺伝子に対するGO解析結果をドットプロットで示しました。縦軸は各GOターム、横軸は各GOタームを持つ遺伝子の内、DEGに含まれていた遺伝子の割合を示します。ドットの大きさは発現変動遺伝子に含まれていた各GOタームを持つ遺伝子の数、ドットの色は調整済みp値を示します。Up側では代謝に関連するGOターム、減少側では血球細胞分化に関連するGOタームが多く含まれていました。
階層性を考慮したGO解析結果の可視化
階層性を考慮したGO解析結果の可視化
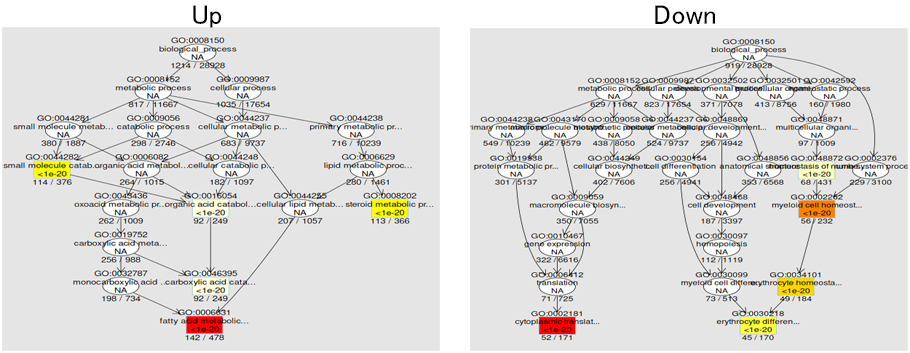
GOタームには階層性があり、広い概念を包括するGOタームは、より狭い概念のGOタームを含んでいます。図の上部に広い概念、下部に狭い概念のGOタームを配置してGO解析の結果を可視化しました。各GOタームの背景色は白色>黄色>オレンジ>赤色の順に有意確率が低くなり、統計的により信頼性が高い事を意味します。図の下側のGOタームに濃縮が見られた場合には、より特異的な生物学的応答が起こったと推定されます。
2群間発現比較におけるGSEA結果
2群間発現比較におけるGSEA結果
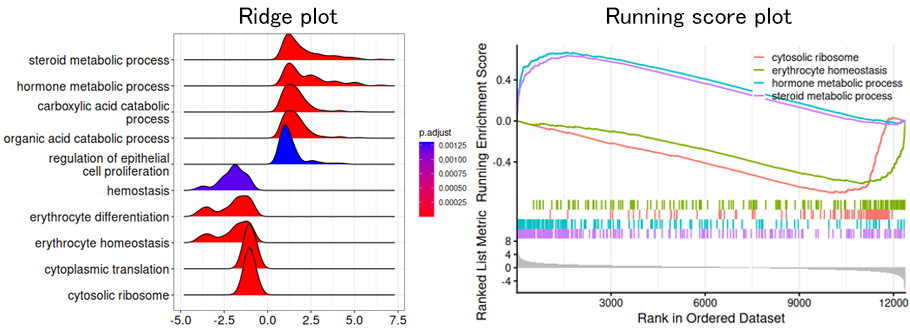
特定の生命現象に関連する遺伝子のセット(gene set)に含まれる遺伝子が、出生後の遺伝子発現変化で増加・減少の一方に濃縮されていた遺伝子セットをGSEAにより抽出しました。day10での増加側および減少側に濃縮が見られた遺伝子セットからそれぞれ5セットを選択し、 Ridge plot(左図)で、上から正規化エンリッチメントスコア(NES)の高い順にプロットしました。各プロットの縦軸はその発現変動比に分布する遺伝子の確率分布(割合に相当)、横軸は発現変動比(log2)を示します。各遺伝子セットのNESに寄与した遺伝子の発現変動比の分布を概観できます。Running score plot(右図)で増加側と減少側の各2遺伝子セットを色分けして表示しました。縦軸は濃縮のスコア、横軸は各遺伝子の発現変動比の大きい順に左から右に並べた順位を示します。
解析例2 幹細胞から分化誘導した心房細胞の薬剤処理による遺伝子発現変化
幹細胞の利用方法の一つとして、多能性幹細胞を分化誘導した細胞を用いた創薬スクリーニングの高効率化が期待されています。ibrutinibは有用な経口白血病治療薬ですが、心房細動を増加させる事が知られています。ヒト多能性幹細胞から誘導した心房心筋細胞をibrutinib処理し、RNA-seq解析を実施したデータを使用して、 ibrutinib処理による心房心筋細胞のRNA発現変化を検出し、発現変動遺伝子が多く含まれていたパスウェイを抽出しました。
遺伝子発現量推定結果
遺伝子発現量推定結果
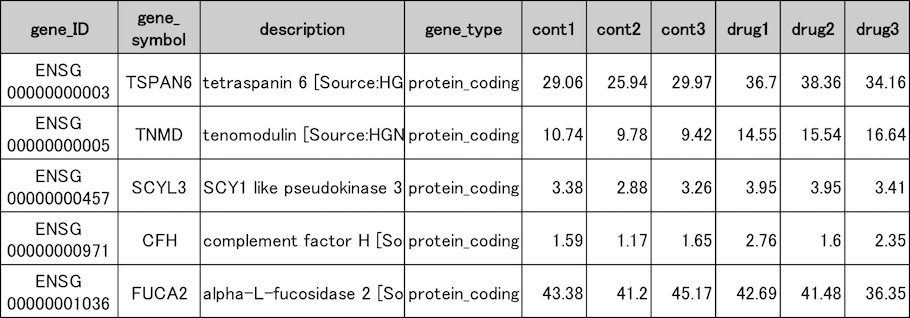
薬剤未処理の(cont1〜3)心房心筋細胞と薬剤処理した細胞(drug1〜3)の遺伝子発現量推定結果の一部を示しました。
発現変動遺伝子抽出結果(抜粋)
発現変動遺伝子抽出結果(抜粋)
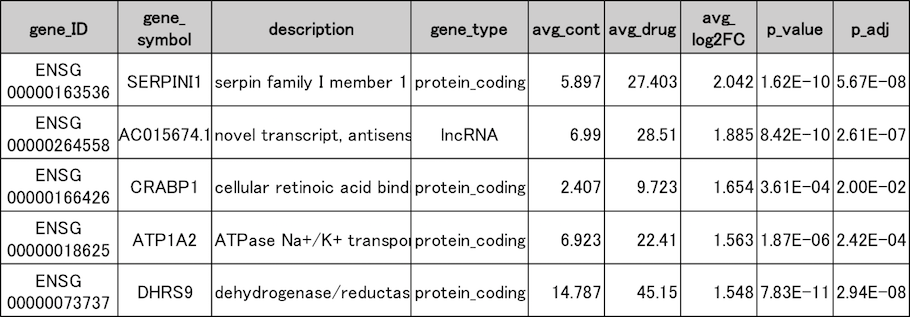
薬剤未処理群(cont1〜3)と処理群(drug1〜3)の発現変動遺伝子の一部を抜粋して示しました。
発現変動遺伝子の可視化(Heatmap)
発現変動遺伝子の可視化(Heatmap)
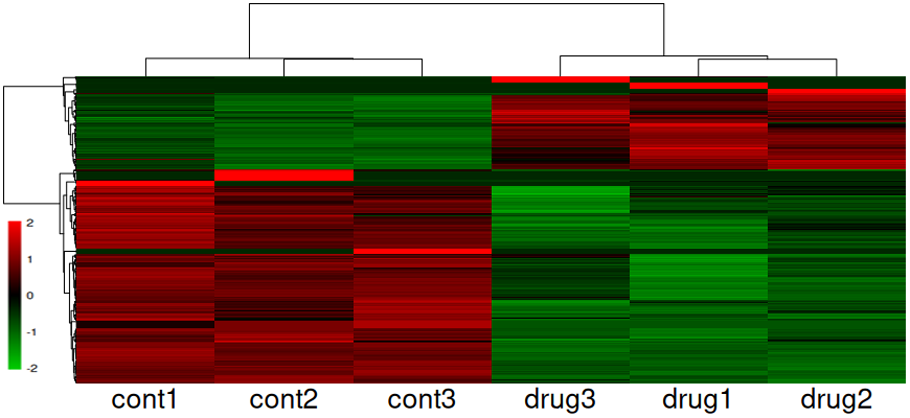
発現変動遺伝子の発現変化をヒートマップで一覧表示しました。各サンプルの発現量をZ-scoreで標準化し、発現量が多い遺伝子を赤色、少ない遺伝子を緑色、中間を黒色で表示しました。図の縦軸は遺伝子、横軸はサンプルを表します。
発現変動遺伝子の可視化(Volcano Plot)
発現変動遺伝子の可視化(Volcano Plot)
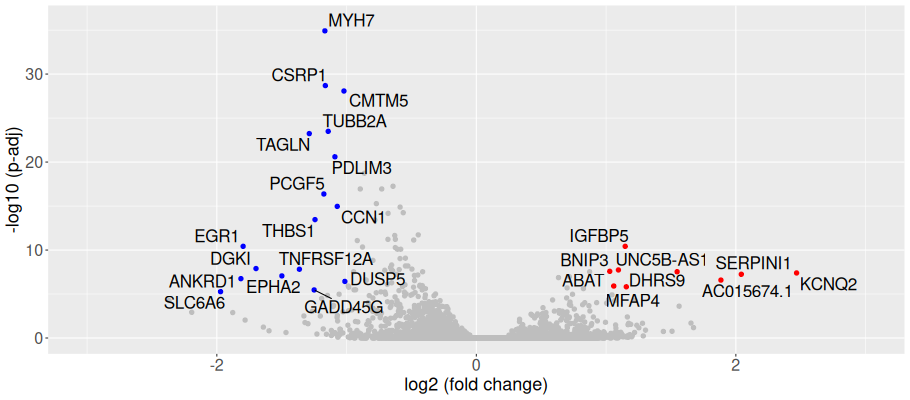
ボルケーノプロットで発現変動比と統計的有意差を一覧表示しました。各点は遺伝子を表しています。発現変動比と調整済みp値によって分類し色分け表示しました。縦軸は調整済みp値を-log10、横軸は発現変動比をlog2で示しています。
発現変動遺伝子のパスウェイ解析
発現変動遺伝子のパスウェイ解析
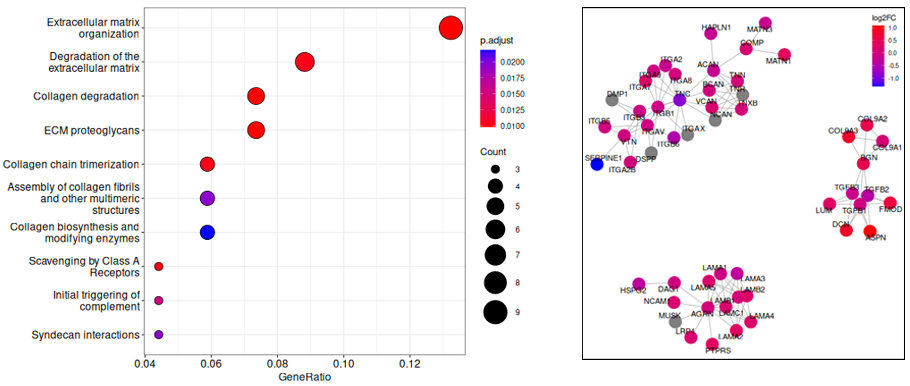
発現変動遺伝子が濃縮されていたパスウェイを抽出し、ドットプロット(左図)で示しました。縦軸はパスウェイ、横軸はパスウェイを構成する全遺伝子に対して、含まれていた発現変動遺伝子の割合を示します。ドットのサイズはパスウェイを構成する遺伝子数、色は補正済p値を示します。右図は“ECM proteoglycans”に含まれる遺伝子と薬剤処理による発現量の増減を示しています。ドットはパスウェイに含まれる遺伝子を示し、発現が増加した遺伝子を赤、減少した遺伝子を青で示しました。
解析例3 がん浸潤T細胞サブタイプの発現変動遺伝子抽出
大腸がん患者から採取したがん浸潤CD8+ T細胞と、末梢血中のCD8+ T細胞をFACSによりソーティングしたT細胞サブタイプのRNA-seq解析データを用いて、それぞれのサブタイプに特徴的な発現変動遺伝子を抽出しました。さらに末梢血のCD8+ T細胞とがん応答性CD8+ T細胞サブタイプ間の発現変動遺伝子が濃縮していたパスウェイを抽出し、パスウェイに含まれる遺伝子の発現変化を可視化しました。
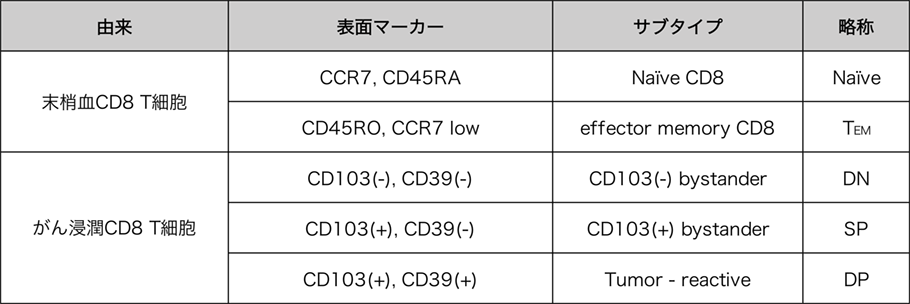
既知の細胞表面マーカーによるCD8+ T細胞の分類
既知の細胞表面マーカーによるCD8+ T細胞の分類
各CD8+ T細胞サブタイプに特徴的な遺伝子発現(ヒートマップ)
各CD8+ T細胞サブタイプに特徴的な遺伝子発現(ヒートマップ)
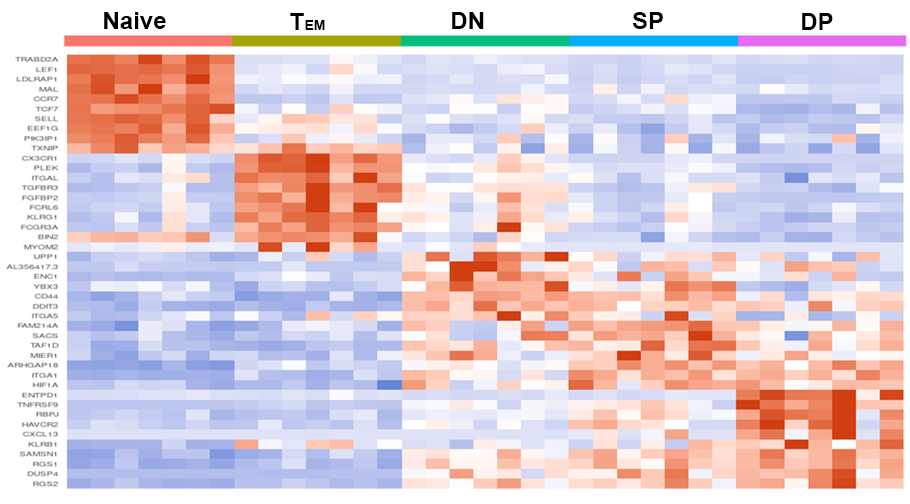
各CD8+ T細胞サブタイプに特徴的な遺伝子の発現をヒートマップで一覧表示しました。FACSによりソーティングされた集団ごとの特徴が網羅的に抽出されました。
PCAおよび相関分析による各検体間の類似性評価
PCAおよび相関分析による各検体間の類似性評価
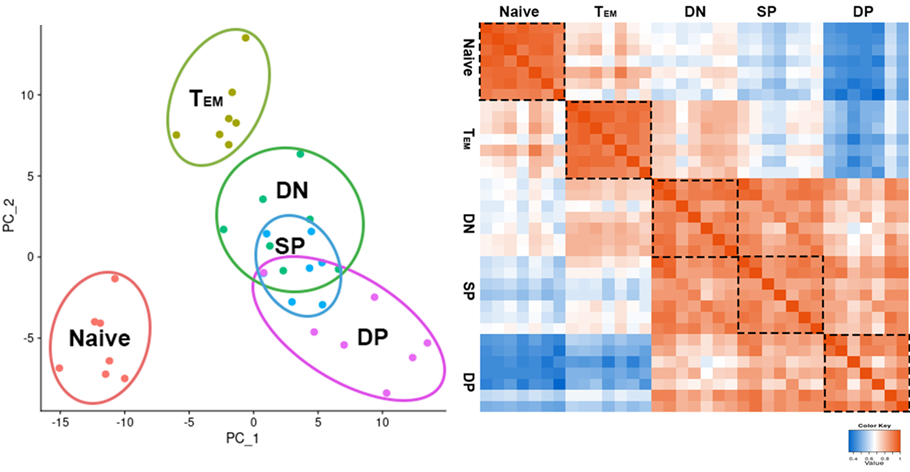
PCA(主成分分析、左図)および、検体間の相関係数のヒートマップ表示(右図)により各検体間の類似性を評価しました。既知の細胞表面マーカーで明確に分類できない検体については、1細胞ごとにRNA-seqデータを取得するシングルセルRNA-seqによる詳細な解析が有効です。
がん浸潤CD8+ T細胞のサブタイプごとの発現変動遺伝子のベン図
がん浸潤CD8+ T細胞のサブタイプごとの発現変動遺伝子のベン図
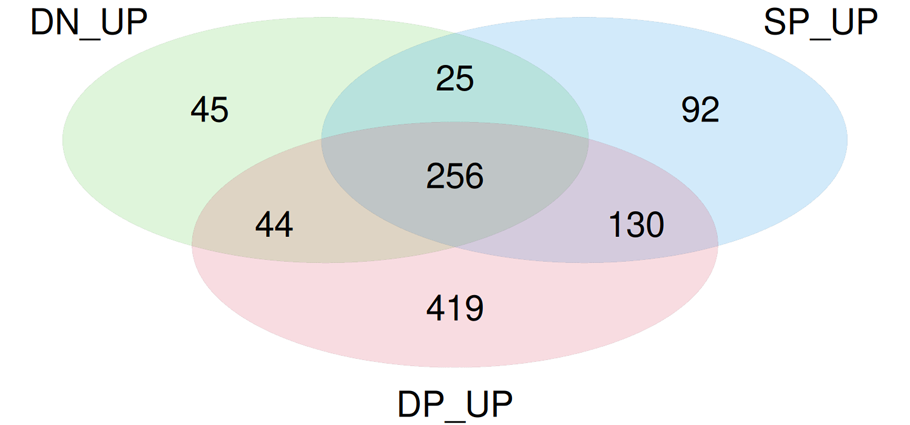
3種類のがん浸潤CD8+ T細胞サブタイプとTEM細胞の発現プロファイルを比較して、それぞれのがん浸潤CD8+ T細胞サブタイプで発現量が増加した発現変動遺伝子を抽出し、その集合関係をベン図で表示しました。
発現変動遺伝子の可視化
発現変動遺伝子の可視化
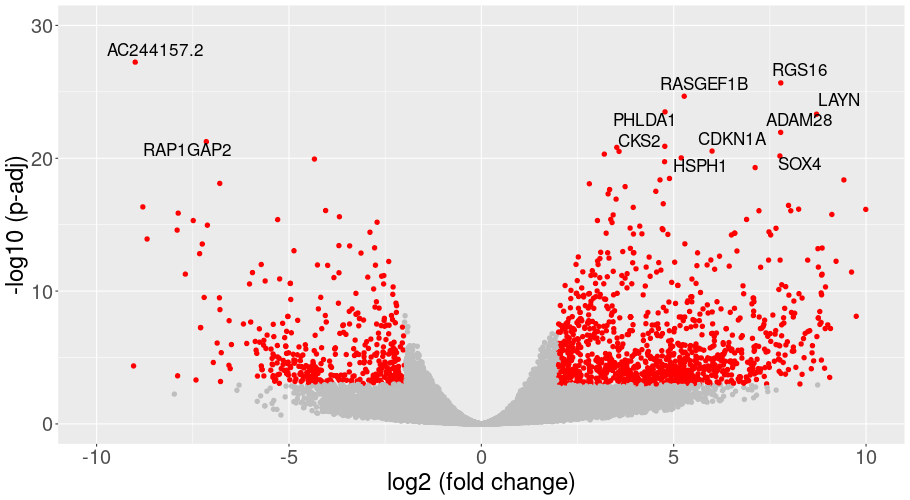
DP(がん応答性サブタイプ)とTEMの間で発現が変化した遺伝子を、発現変動比と統計的有意差を用いたボルケーノプロットで一覧表示しました。各点は遺伝子を表しています。
発現変動遺伝子のパスウェイ解析
発現変動遺伝子のパスウェイ解析
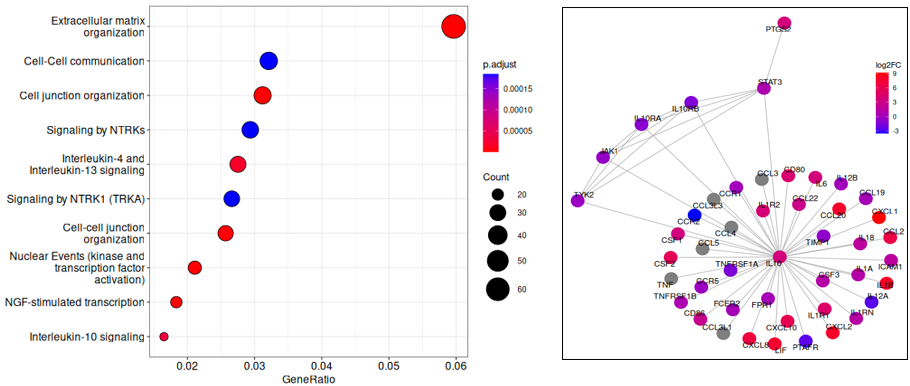
DPとTEMの間の発現変動遺伝子が濃縮されていたパスウェイを抽出し、ドットプロット(左図)で示しました。縦軸はパスウェイ、横軸はパスウェイを構成する全遺伝子に対して、含まれていた発現変動遺伝子の割合を示します。ドットのサイズはパスウェイを構成する遺伝子数、色は補正済p値を示します。右図は“Interleukin-10 signaling”に含まれる遺伝子と発現量の増減を示しています。ドットはパスウェイに含まれる遺伝子を示し、 DPで発現が増加した遺伝子を赤、減少した遺伝子を青で示しました。
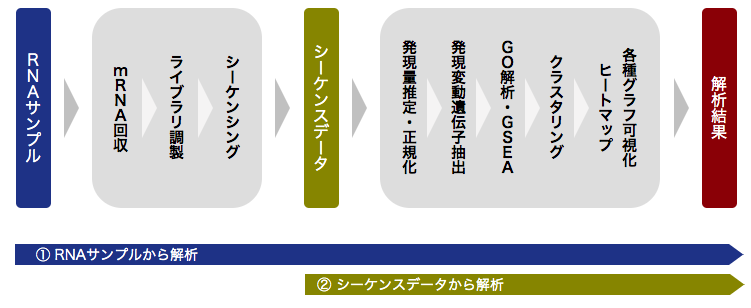
解析フロー
組織・RNA
mRNA回収
ライブラリ調製
シーケンシング
シーケンスデータ
発現量推定・正規化
クラスタリング
発現変動遺伝子抽出
遺伝子機能解析
高次解析
解析結果
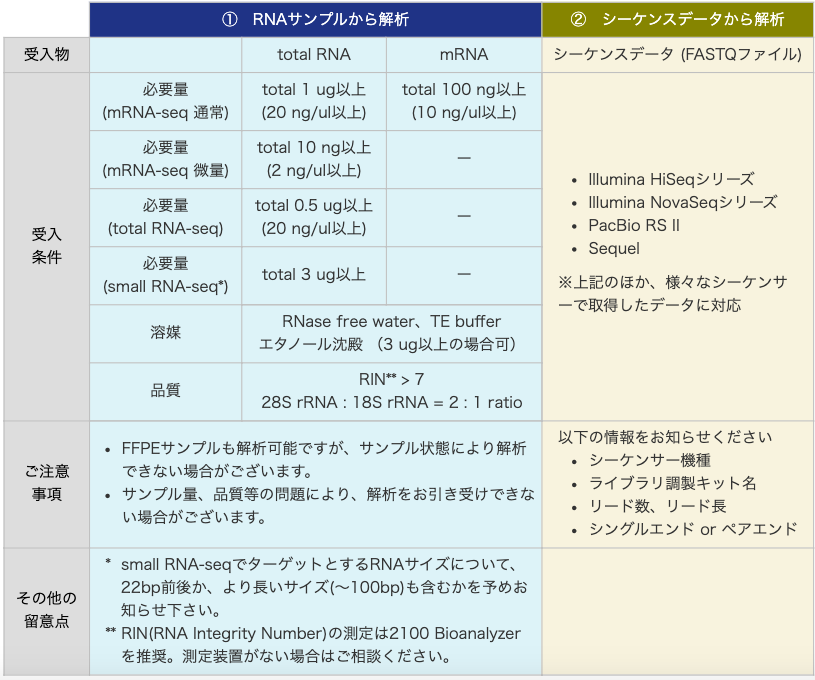
受入物
| ① 組織 / RNAから解析 | ② シーケンスデータから解析 | |||
|---|---|---|---|---|
| 受入物 | total RNA | mRNA | シーケンスデータ (FASTQファイル) | |
| 受入 条件 |
mRNA-seq 通常量タイプ |
total 1 ug以上 (20 ng/ul以上) |
total 100 ng以上 (10 ng/ul以上) |
※上記のほか、様々なシーケンサーで取得したデータに対応
|
| mRNA-seq 微量タイプ |
total 10 ng以上 (2 ng/ul以上) |
ー | ||
| total RNA-seq | total 0.5 ug以上 (20 ng/ul以上) |
ー | ||
| small RNA-seq* | total 3 ug以上 |
ー | ||
| 溶媒 | RNase free water エタノール沈殿 (3 ug以上の場合可) |
|||
| 品質 | RIN** > 7 28S rRNA : 18S rRNA = 2 : 1 ratio |
|||
| ご注意 事項 |
|
以下の情報をお知らせください
|
||
| その他の 留意点 |
|
|||
解析内容
- mRNAシーケンシング(通常量・微量タイプ)
-
- 網羅的なmRNAの塩基配列決定
- mRNAの網羅的な発現量推定、発現差解析に最適(別途データ解析)
-
【 解析内容 】
- 検体の受入検査(RNA量、濃度、純度、分解の有無)
- ライブラリ調製
- シーケンシング
-
【 解析条件 】
- 検体受入検査 : Agilent Bioanalyzer / TapeStation
- シーケンサー : Illumina Novaseq 6000 / X
- シーケンス : 100bp、ペアエンド、4,000万リード(4Gb/検体)
- Total RNAシーケンシング
-
- 網羅的なmRNA、lncRNA等のnoncoding RNAの塩基配列決定
- 分解が進んだtotal RNAからのmRNAの塩基配列決定
- mRNA、lncRNA等のnoncoding RNAの発現量推定、発現差解析に最適(別途データ解析)
-
【 解析内容 】
- 検体の受入検査(RNA量、濃度、純度、分解の有無)
- rRNA除去
- ライブラリ調製
- シーケンシング
-
【 解析条件 】
- 検体受入検査 : Agilent Bioanalyzer / TapeStation
- シーケンサー : Illumina Novaseq 6000 / X
- シーケンス : 100bp、ペアエンド、4,000万リード(4Gb/検体)
- データ解析(スタンダード1)
-
- 発現量推定が目的で、安価に解析したい場合にお勧めです。
- k-mer countingにより発現量を推定
- 既知遺伝子、転写産物の発現量推定
- マッピングの可視化が不要な場合に最適
- 他の解析でBAMファイルを使用しない場合に推奨
- mRNAおよびTotal RNAシーケンシングのデータに対応
- 参照配列はトランスクリプトームを使用
-
【 解析内容 】
- データ受入検査(データ量、シーケンス品質、アダプタ配列の有無等)
- 不要な配列のトリミング
- k-mer counting
- 発現量推定・正規化
- 発現変動遺伝子抽出(DEG抽出、発現比較解析)
- データ解析(スタンダード2)
-
- 発現量推定が目的で、マッピングの可視化が必要な場合にお勧めです。
- 参照ゲノム配列にマッピングし発現量を推定
- 既知遺伝子、転写産物の発現量推定
- 他の解析でBAMファイルを使用する場合に推奨
- mRNAおよびTotal RNAシーケンシングのデータに対応
- 参照配列はゲノムを使用
-
【 解析内容 】
- データ受入検査(データ量、シーケンス品質、アダプタ配列の有無等)
- 不要な配列のトリミング
- マッピング
- 発現量推定・正規化
- 発現変動遺伝子抽出(DEG抽出、発現比較解析)
※その他、各種解析やグラフの作成も承っております。
・PCA(主成分分析)
・GO解析
・GSEA
・パスウェイ解析
・共発現ネットワーク解析
・各種ヒートマップ
・Volcano plot
・ベン図
価格・納期
ご要望に合わせて実験デザインをご提案し、御見積致しますのでこちらよりお問い合わせ下さい。
※全解析(ウェット+ドライ)のご依頼の場合は、6検体から受け入れています。
※ウェットのみのご依頼の場合は、12検体から受け入れています。
ご注意事項
受入物を品質検査した結果、解析をお引き受けできない場合があります。
お問い合わせ
こちらよりお問い合わせください。
専門技術者が原則24時間以内にご連絡します。(土日祝日は除く)
参考文献
Sumedha S. Gunewardena et al. “Deciphering the Developmental Dynamics of the Mouse Liver Transcriptome.” PLoS One. 2015 Oct 23;10(10) e0141220. doi: 10.1371/journal.pone.0141220.
Shafaattalab, S. et al. “Ibrutinib Displays Atrial-Specific Toxicity in Human Stem Cell-Derived Cardiomyocytes.” Stem Cell Reports 12, 996–1006 (2019). doi: 10.1016/j.stemcr.2019.03.011.
Yang, R. et al. “Distinct epigenetic features of tumor-reactive CD8+ T cells in colorectal cancer patients revealed by genome-wide DNA methylation analysis.” Genome Biology 21, (2020). doi:10.1186/s13059-019-1921-y
*1 Gene Ontology
*2 Gene Set Enrichment Analysis