
RamDA-seq / Shin-RamDA-seq解析
1細胞完全長total RNA-seq解析(RamDA-seq®*1 / Shin-RamDA-seq®*1)とは
1細胞ごとに、polyA RNAおよびnon-polyA RNAの種類と量を網羅的に検出し、かつRNAの全長を偏りなく検出することが可能です。発現プロファイルを基に細胞集団を分類し、亜集団における検体間の多種多様なRNA量の変動を検出することができます。lncRNAやpre-mRNA等の特殊なRNAの検出も可能です。
RNAには3’末端にpolyA 配列を持つpolyA RNAと、持たないnon-polyA RNAがあります。これまで従来のシングルセルRNA-seq解析法ではnon-polyA RNAは検出できませんでしたが、RamDA-seq解析法ではpolyA RNAだけでなくnon-polyA RNAの両方のRNA種を、1細胞単位で検出することが可能です。また、RNAの長さに依存することなく、各RNA種の5’領域から3’領域に渡る全長を偏りなく高感度に検出することも可能です。
近年、non-polyA RNAを含むnon-coding RNAが生命機能や疾患に関与していることが明らかとなり、その重要性が注目されています。non-polyA RNAを含む各RNA種の発現プロファイルを1細胞レベルで解析することで、新たな知見が得られることが期待されます。
当データ解析サービスは、理化学研究所 生命機能科学研究センター バイオインフォマティクス研究開発チーム(チームリーダー二階堂愛博士)の技術指導および監修の下にご提供しています。
用途
- 1細胞レベルでのcodingおよびnon-coding RNA(lncRNAなど)の発現解析
- 超微量RNA(10pg~)からのtotal RNAシーケンス解析
- 1細胞レベルのスプライシング異常検出
- 細胞集団に含まれる各細胞タイプの推定*3(未知の細胞タイプ検出を含む)
- 細胞集団に含まれる各細胞タイプで特徴的に発現する遺伝子の同定
- 群間における各細胞タイプでの発現比較(発現変動遺伝子の高感度検出)
- 多数の細胞中に混在する希少細胞における高感度な発現変化の検出
- 擬時間(pseudotime)解析による高精度な分化経路推定
※関連する解析サービス
従来法(polyA RNA検出)のシングルセルRNA-seq解析 >
ホルマリン固定後のシングルセルトランスクリプトーム解析 >
シングルセルレパトア解析+シングルセルRNA-seq解析 >
シングルセル空間トランスクリプトーム解析 >
スポット型空間トランスクリプトーム解析 >
バルクRNA-seq解析 >
このような方に
- 従来法では検出できなかった多くのnon-coding RNA(lncRNAなど)やpre-mRNAを解析したい方
- 従来法では検出が困難な超微量RNA(10pg~)からの遺伝子発現解析をご希望の方
- 自身の研究目的に最適な実験デザインの検討から始めたい方
- 共同研究先の対応スピードや、一方向的な情報科学的解析手法にお悩みの方
当社の特徴
- 共同研究タイプの受託解析サービス(定型的な受託解析ではありません)
- ウェット実験とデータ解析の両分野の経験豊富な専門技術者が直接対応
- RamDA-seq開発元の理研より唯一技術導入した信頼性の高いデータ解析法
- 生物学的意義を踏まえたデータ解析サービス(詳細はこちら)
- 解析結果の生物学的解釈のサポート
従来法との比較(イメージ図)*2
従来のシングルセルRNA-seq解析法ではpolyA RNAの一部の領域しか検出できませんでしたが、RamDA-seqではpolyA RNAおよびnon-polyA RNAの全RNA種を検出可能です。また、各RNA種の5’領域から3’領域に渡る全長の領域(Exon + Intron)を高感度に検出することが可能です。
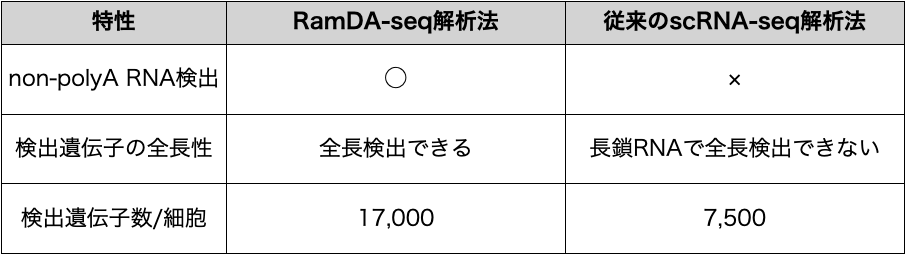
| 特性 | RamDA-seq解析法 | 従来のscRNA-seq解析法 |
|---|---|---|
| polyA RNA検出 | ○ | ○ |
| non-polyA RNA検出 | ○ | × |
| 検出遺伝子の全長性 | 全長検出できる | 長鎖RNAで全長検出できない |
| 検出遺伝子数/細胞 | 17,000 | 7,500 |
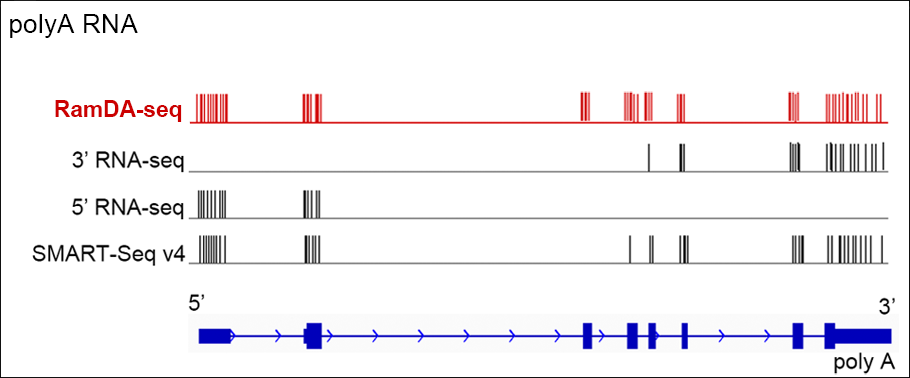
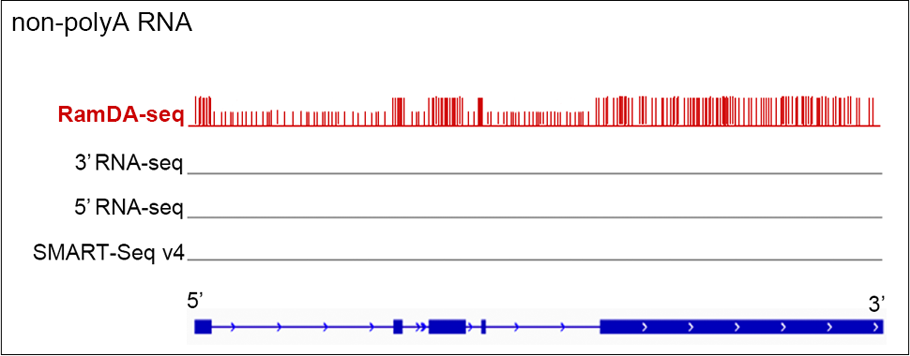
(※) polyA RNAは長鎖mRNAのイメージ図です。
Coding RNAおよびnon-coding RNAにおける検出比較
RNAは、タンパク質に翻訳されるmRNA等のcoding RNAと、翻訳されないlncRNA等のnon-coding RNAに分けられます。従来法では、coding RNAやnon-coding RNAのごく一部しか検出できませんでした。一方 RamDA-seq法では、 coding RNAとnon-coding RNAの両方のRNA種を網羅的にかつ高感度に検出することが可能です。
| coding RNA | non-coding RNA | ||
|---|---|---|---|
| 従来法 | polyA RNA |
・mRNA |
・lncRNA |
| RamDA seq |
・mRNA(高遺伝子検出数) |
・lncRNA(高遺伝子検出数) |
|
| non polyA RNA |
・pre-mRNA(mRNA前駆体) ・histone mRNA |
・lncRNA(高遺伝子検出数) ・enhancer RNA(※1,2) ・circRNA(※2) ・antisense RNA(※3) ・arcRNA(※1,3) ・snoRNA ・snRNA(※1) ・他のnon-coding RNA |
|
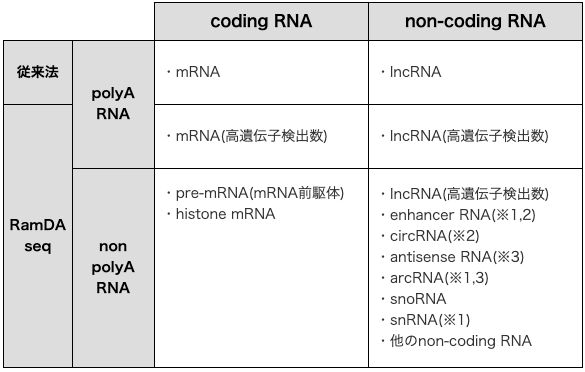
(※1) polyA RNAを一部含む
(※2) lncRNAの1種
(※3) lncRNAを含む
(注1) 主要なRNA種のみ表記しています。
(注2) RNAの分類は表記当時の解釈であり今後追加修正の可能性があります。
解析例1 自己免疫疾患に関与するAire欠損による遺伝子発現変化の検出
機能異常により自己免疫疾患を引き起こすAire遺伝子の欠損による遺伝子発現変化を検出しました。胸腺髄質上皮細胞で発現するAireは組織特異的抗原の異所的な発現を制御しており、自己抗原に反応するT細胞の除去に必須です。Aireによる直接的な遺伝子発現制御の他に、Aireを介した胸腺細胞の分化成熟による間接的な遺伝子発現変化があると考えられています。Aire発現の有無による直接的、および間接的な遺伝子発現変化を検出しました。
野生型(WT)およびAire欠損(KO)胸腺細胞のクラスタリング結果
野生型(WT)およびAire欠損(KO)胸腺細胞のクラスタリング結果
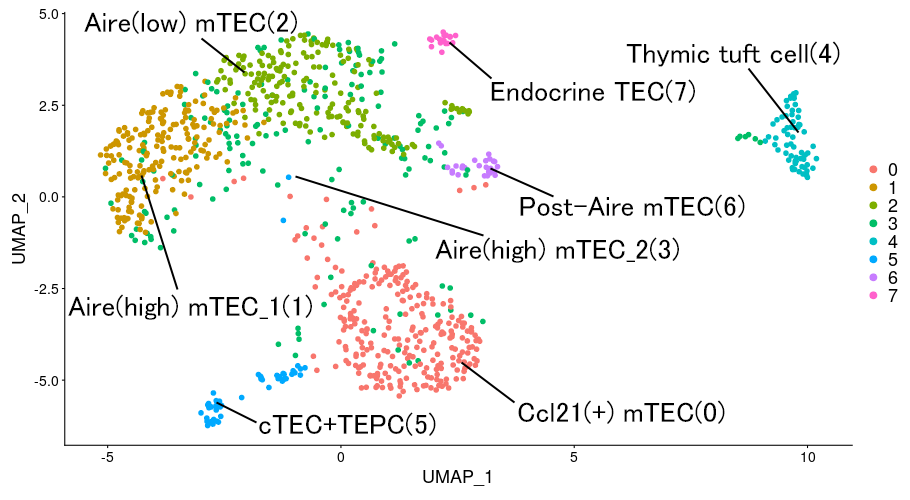
野生型(WT)およびAire欠損(KO)胸腺細胞の発現プロファイルをBatch effect(実験間誤差)補正後に統合しクラスタリングした結果を、UMAPにより二次元上にプロットしました。細胞タイプ名に続く()内の数字はクラスタ番号を示します。mTEC: medullary thymic epithelial cell, cTEC: cortical TEC, TEPC: thymic epithelial progenitor cell.
WTおよびKOにおけるAireの発現分布と発現レベル
WTおよびKOにおけるAireの発現分布と発現レベル
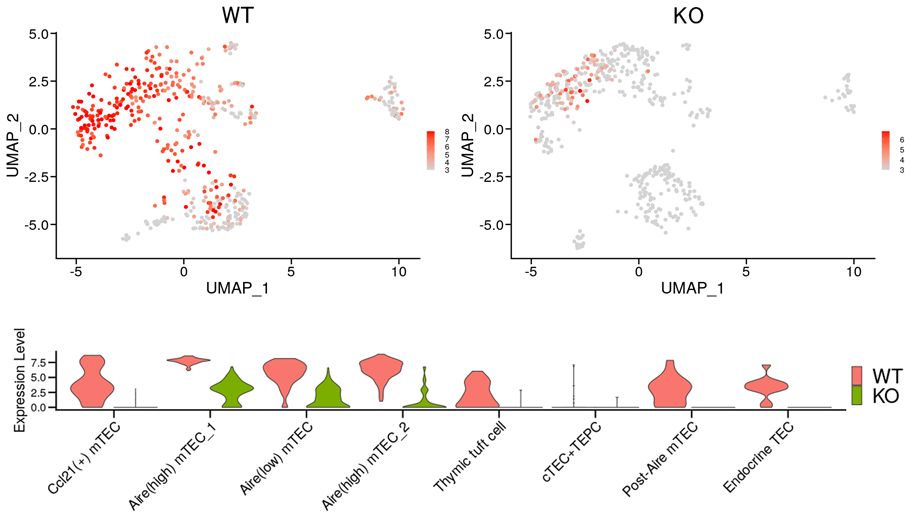
Aireの発現分布をWTとKO(上段左:WT、右:KO)に分けて図示し、発現量が多い細胞を赤色で示しました。各細胞タイプのAire発現量をバイオリンプロット(下段)で示しました。縦軸は発現量、横軸は細胞タイプを示しており、WTを左側、KOを右側に並べて表示しました。Aire(high) mTEC_1、 Aire(high) mTEC_2ではほとんどの細胞でAireの強い発現が見られ、KOでは顕著な発現量の減少が見られました。
野生型(WT)およびAire欠損(KO)胸腺細胞の分布と各クラスタの細胞構成比
野生型(WT)およびAire欠損(KO)胸腺細胞の分布と各クラスタの細胞構成比
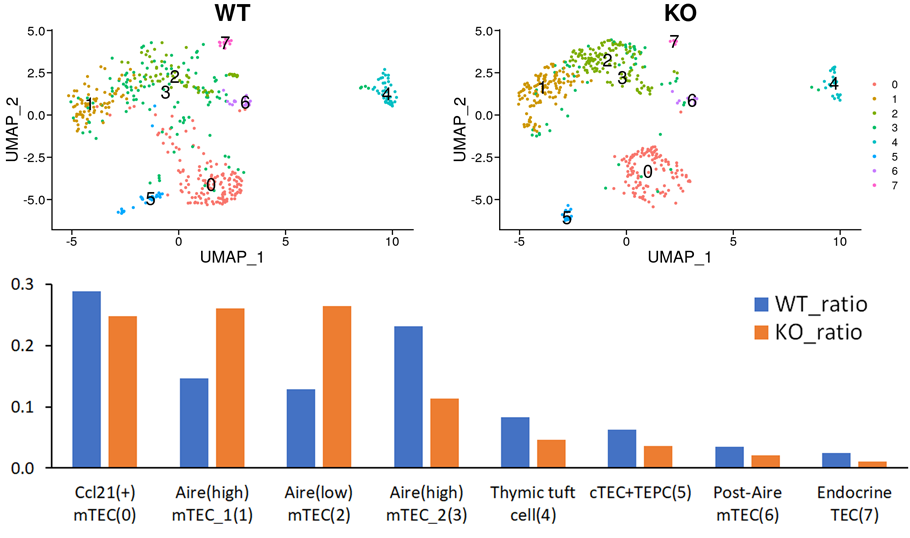
クラスタリング結果を、WT(上段左)およびKO(上段右)に分けて図示し、各細胞タイプを構成するWTとKOの割合をグラフ(下段)で表示しました。グラフ縦軸はWT、KOそれぞれの細胞数の合計を1として各細胞タイプに含まれる細胞の割合を示します。KOではAire(high) mTEC_2の割合が減少し、Aire(high) mTEC_1とAire(low) mTECの割合が増加する傾向が見られました。
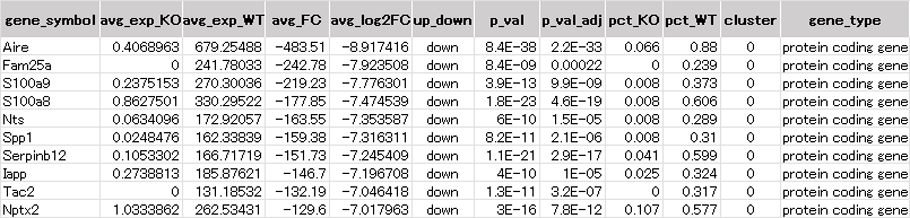
2群間の発現変動遺伝子(DEG)抽出結果
2群間の発現変動遺伝子(DEG)抽出結果
coding RNA
coding RNA
non-coding RNA
non-coding RNA
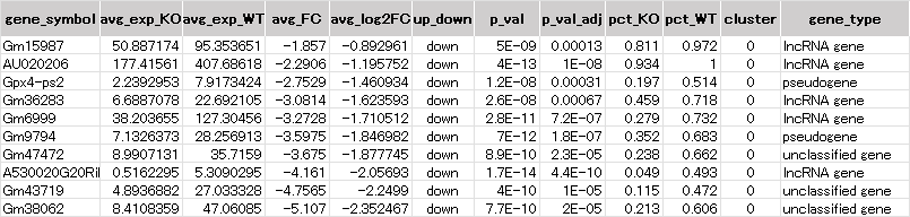
KOにより発現量が減少した発現変動遺伝子をcoding RNA(上段)およびnon-coding RNA(下段)に分けてリスト表示しました。
KOにより発現が減少した発現変動遺伝子(coding RNA)
KOにより発現が減少した発現変動遺伝子(coding RNA)
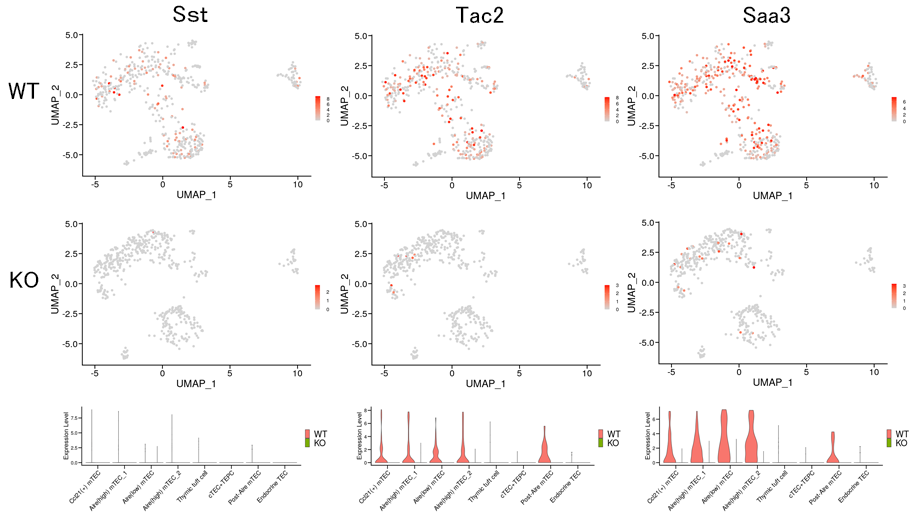
KOにより発現量が減少した発現変動遺伝子の発現分布を群ごと(上段:WT、中段:KO)に分けて図示しました。細胞タイプ毎の遺伝子発現量をバイオリンプロット(下段)で示しました。本来は内分泌細胞に発現するSstや神経細胞に発現するTac2は、同じ細胞タイプの中でも一部の細胞でのみ高い発現を示し、KOにより顕著に発現が減少しました。本来胸腺上皮細胞で発現しない遺伝子が一部の細胞でのみ発現するのはAireにより直接制御される遺伝子の特徴とされています。免疫応答に関係するSaa3は、mTECの大半の細胞で発現が見られ、KOにより発現が顕著に減少しました。AireのKOによる胸腺細胞の分化抑制に起因する可能性が考えられます。
KOにより発現が減少した発現変動遺伝子(non-coding RNA)
KOにより発現が減少した発現変動遺伝子(non-coding RNA)
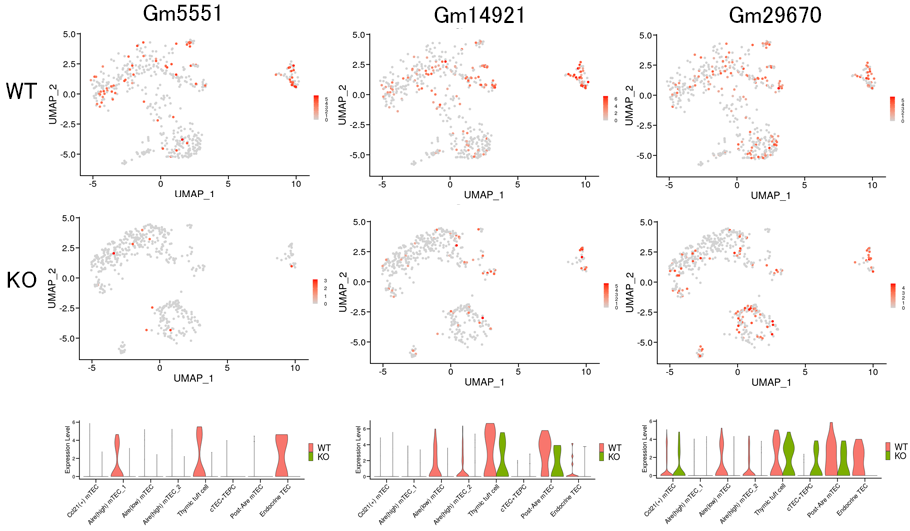
KOにより発現量が減少したnon-coding RNAの発現分布(上段WT、中段KO)、および発現量(下段)を図示しました。coding RNAと同様に、同じ細胞タイプの中でも比較的一部のWTでのみ高い発現を示しKOで顕著に発現が減少する遺伝子(Gm5551)や、比較的多くの細胞タイプで全体的に発現し、KOにより特定の細胞タイプで発現が減少する遺伝子(Gm14921, Gm29670)などが見られました。
解析例2 iPS細胞由来神経幹細胞分化に伴い変動するcoding及びnon-coding RNAの検出
ヒトiPS細胞をembryoid bodyに分化させた細胞を浮遊培養し、形成されたニューロスフィアのRamDA-seqデータ(網羅的なRNA発現プロファイル)を用いて、神経幹細胞の分化に伴い特徴的に発現量が変化するcoding RNAおよびnon-coding RNAの検出を行いました。
ヒトiPS細胞由来Neurosphereのクラスタリング結果(664 cells, 1群、UMAP)
ヒトiPS細胞由来Neurosphereのクラスタリング結果(664 cells, 1群、UMAP)
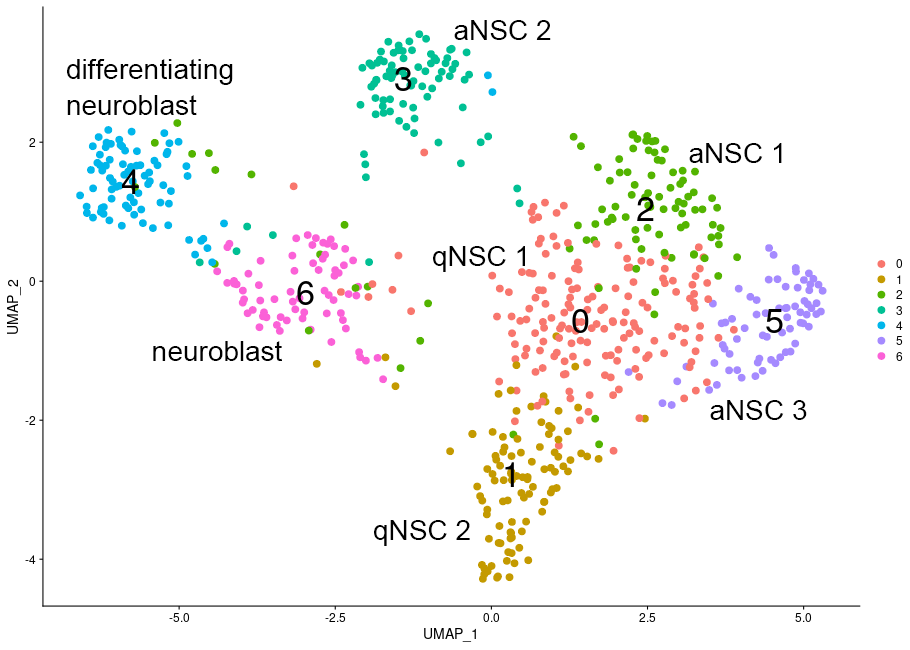
RamDA-seqにより取得された1細胞ごとのcoding RNAおよびnon-coding RNAの両方のRNA種の発現プロファイルに基づいて、細胞集団をクラスタリングしました。増殖中(aNSC)および休止期(qNSC)の神経幹細胞、神経芽細胞(neuroblast)、分化が進行した神経芽細胞(differentiating neuroblast)など複数の細胞タイプに分類されました。
各クラスタに特徴的な遺伝子発現
各クラスタに特徴的な遺伝子発現
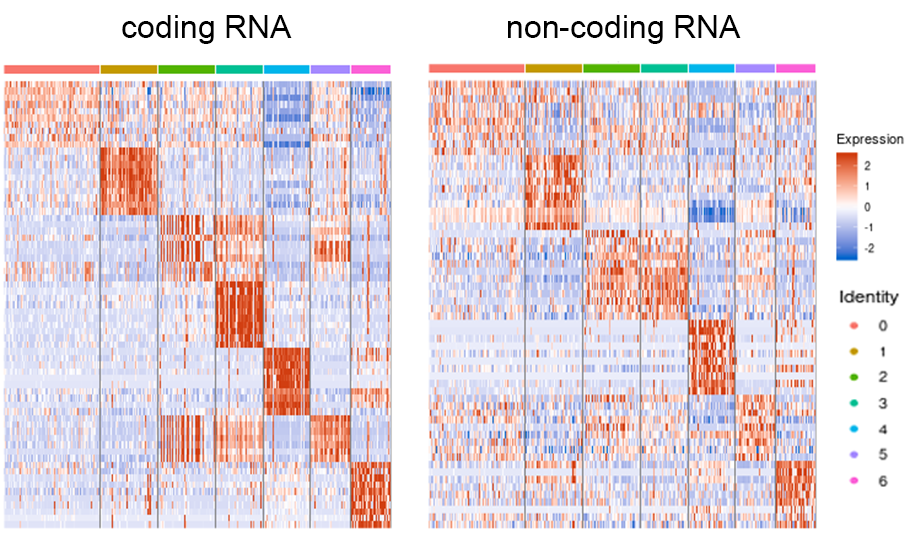
各クラスタに特徴的に発現する遺伝子をヒートマップで一覧表示しました。発現量が多い遺伝子は赤色、少ないものは青色で示しました。
遺伝子発現分布
遺伝子発現分布
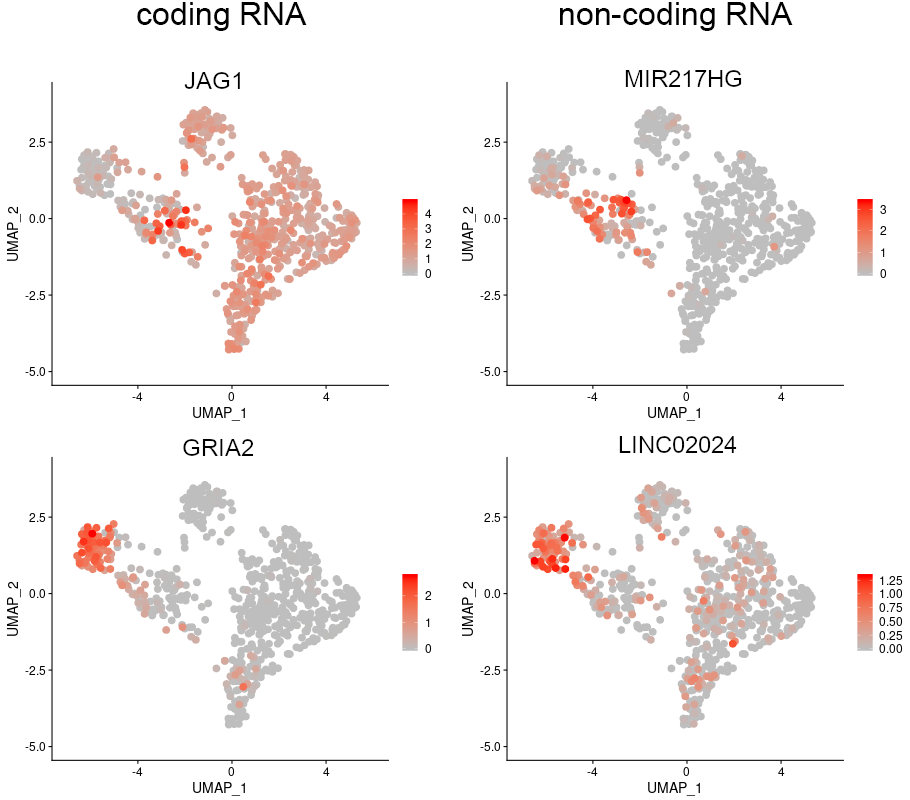
特定のクラスタで特徴的に発現している遺伝子の発現分布を図示しました。 coding RNA、non-coding RNAいずれも特定のクラスタで発現している遺伝子が見られました。MIR217HG : lncRNA(non-polyA RNA)、LINC02024 : lncRNA(lincRNA)
各クラスタにおける遺伝子発現レベル
各クラスタにおける遺伝子発現レベル
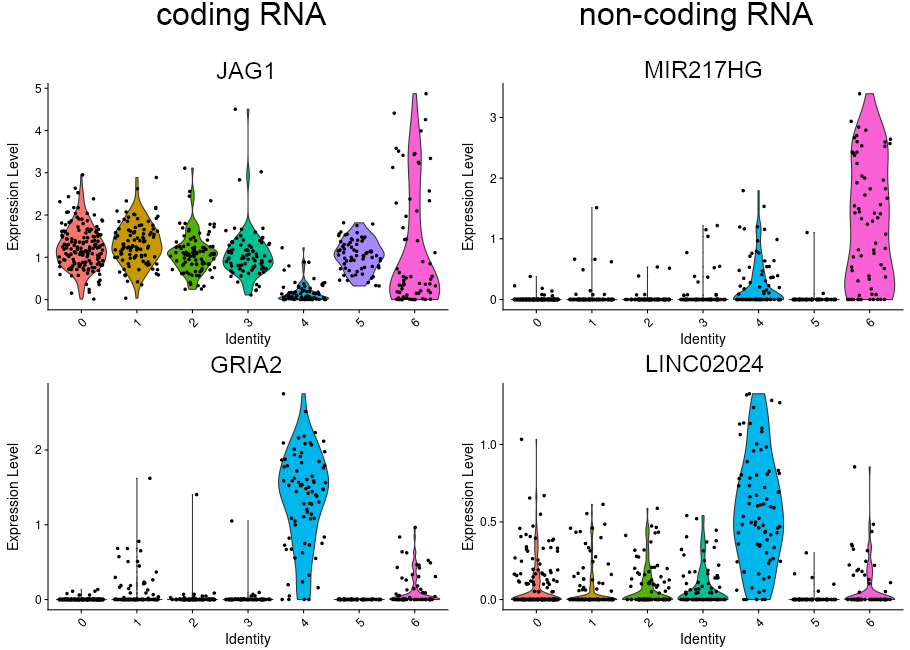
特定のクラスタで特徴的に発現している遺伝子の発現量をバイオリンプロットで図示しました。
細胞ごとのマッピング状態の可視化
細胞ごとのマッピング状態の可視化
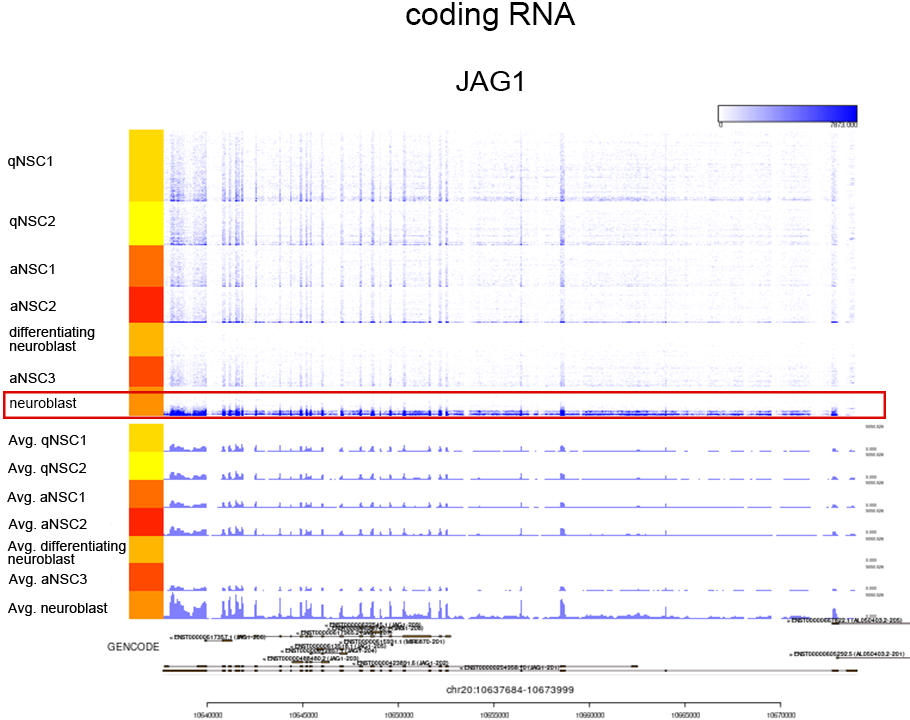
neuroblastで発現が上昇していたJAG1(coding RNA)の細胞ごとのマッピング状態をMillefy*4により可視化しました。neuroblastの一部の細胞(赤枠)ではイントロンを含むJAG1遺伝子全長に渡ってリードがマップされていました。スプライシング 完了前のpre-mRNAが検出されたと考えられます。縦軸は個々の細胞、横軸はゲノム上の位置を示します。
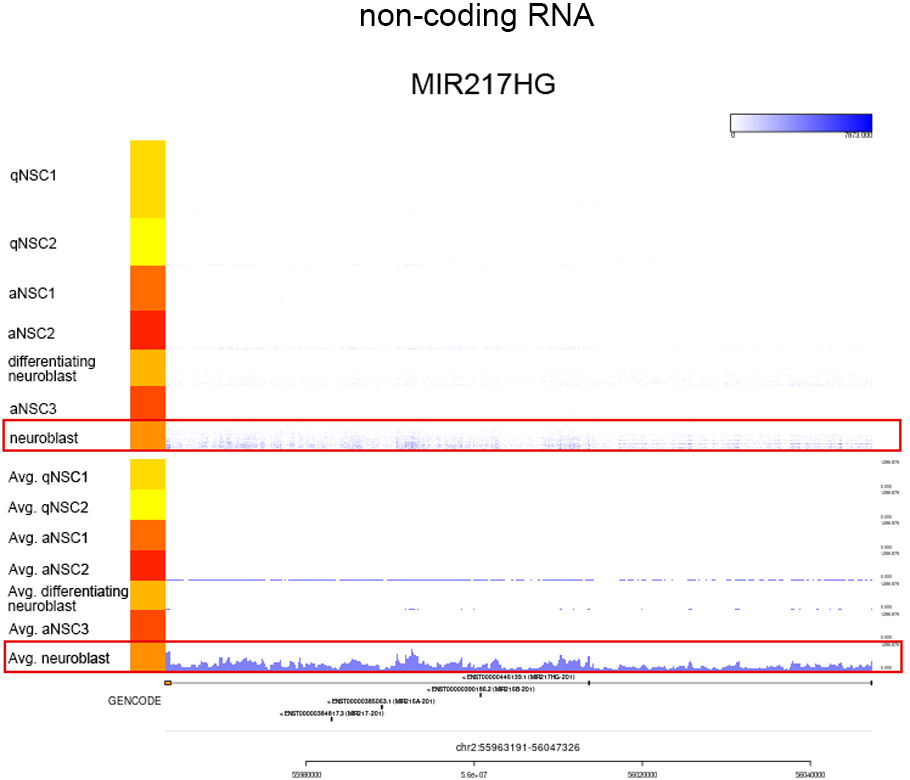
neuroblastで発現上昇していたMIR217HG(lncRNA, non-polyA RNA)のマッピング状態を可視化しました。発現が見られた細胞の多く(上段赤枠内)で、イントロンを含む遺伝子全長に渡ってリードがマップされていました。クラスタ毎の平均値(下段赤枠)からも、遺伝子の領域によらず全長に渡ってリードが取得されたことが示されました。
分化経路推定(trajectory analysis)
分化経路推定(trajectory analysis)
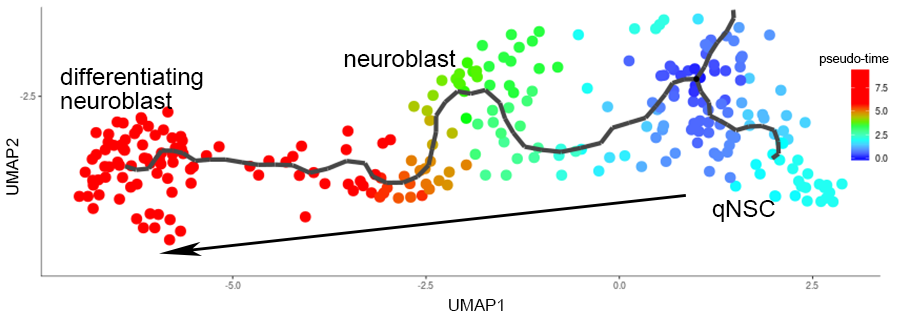
未分化のneural stem cellからneuroblastへの分化経路を推定し、細胞を擬時間で色分け表示しました。矢印は擬時間経過の方向を示しています。
分化経路に沿って特徴的に変化した遺伝子の抽出
分化経路に沿って特徴的に変化した遺伝子の抽出
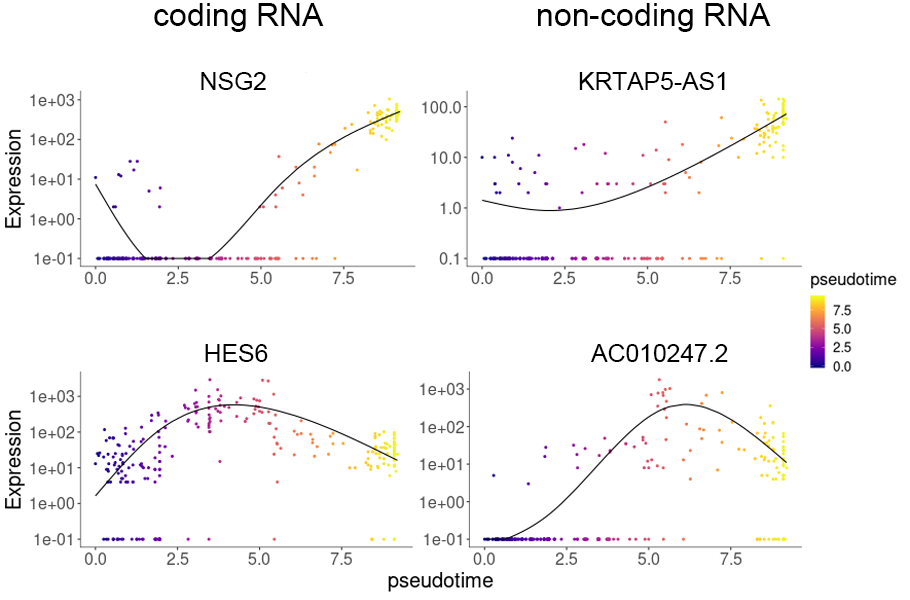
擬時間(pseudotime)に従って特徴的に変化した遺伝子を抽出し、その発現量をグラフ表示しました。縦軸は遺伝子の発現量、横軸は擬時間を表します。分化が進んだ細胞集団で発現上昇する遺伝子(上段)や、分化の過程で一過性に発現上昇する遺伝子(下段)がcoding RNA、non-coding RNAいずれにも見出されました。KRTAP5-AS1 : lncRNA(antisense RNA, non-polyA RNA)、AC010247.2 : lncRNA(antisense RNA, non-polyA RNA)
分化経路に沿って特徴的に変化した遺伝子の発現分布
分化経路に沿って特徴的に変化した遺伝子の発現分布
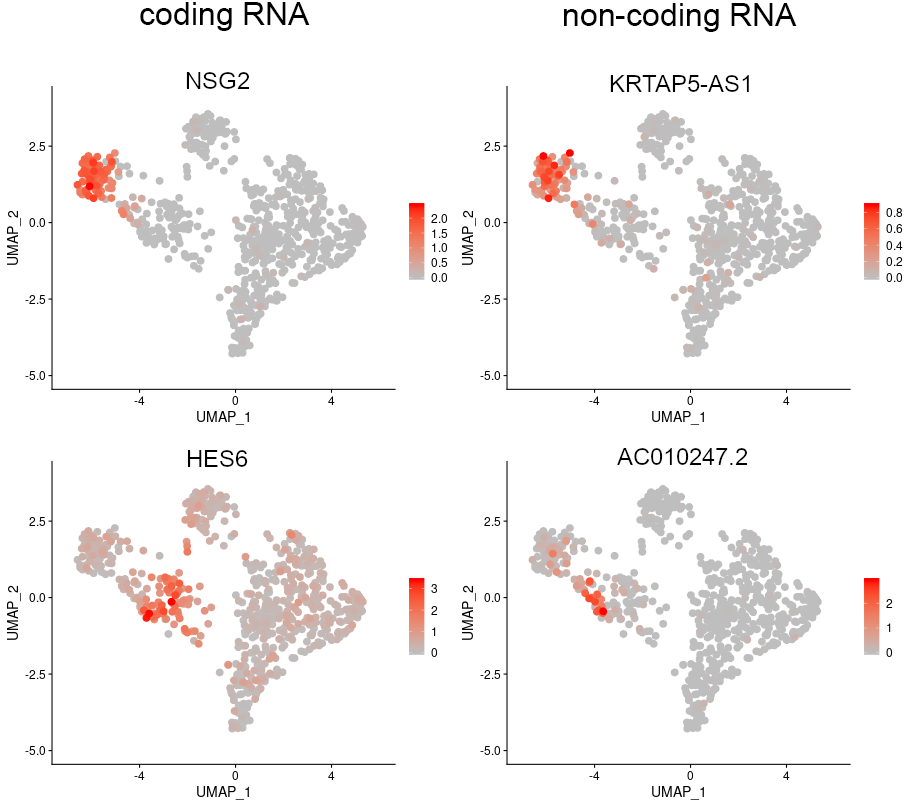
擬時間経過に従って特徴的に発現が変化していた遺伝子の発現分布を示しました。擬時間経過による発現変化に対応する細胞集団で特徴的に発現していることが確認されました。
RamDA-seq法により、近年、疾患との関連が注目されつつあるnon-coding RNAや スプライシング異常を1細胞レベルで検出することが可能になりました。従来のシングルセルRNA-seq法では検出できなかったRNA種やRNA領域で、全く新しい1細胞レベルの知見が得られることが期待されます。
解析フロー
シングルセル化細胞
ライブラリ調製
シーケンシング
シーケンスデータ
発現量推定・正規化
クラスタリング
発現変動遺伝子抽出
細胞タイプ推定
高次解析
解析結果

受入物
| ① シングルセル化細胞から解析 | ② シーケンスデータから解析 | |
|---|---|---|
| 受入物 |
|
|
| 生物種 |
| ― |
| 受入条件 |
※細胞は通常total RNA 10~20pg/細胞程度
|
|
| その他 |
※超微量バルクRNA-seqとしても解析可能
1~100細胞 or total RNA 10pg~1ng/検体 |
以下の情報をお知らせください
|

納品物
①シングルセル化細胞から解析- 解析報告書
- シーケンスデータ
- 細胞クラスタリング画像データ
- 各クラスタの細胞数データ
- 各種遺伝子の発現データ(発現量、発現分布)
- 各種遺伝子リストなど
- 解析報告書
- 細胞クラスタリング画像データ
- 各クラスタの細胞数データ
- 各種遺伝子の発現データ(発現量、発現分布)
- 各種遺伝子リストなど
※実際の納品物は、ご依頼内容により変わります。
価格・納期
ご依頼の解析内容により異なりますので、詳細はこちらよりお問い合わせください。
ご注意事項
受入物を品質検査した結果、解析をお引き受けできない場合があります。
よくあるご質問(FAQ)
- 解析できる生物種について教えてください。
- ヒトとマウスに対応しています。その他の生物種についてはご相談下さい。
- 受託解析サービスの種類について教えてください。
- 現在、「シングルセル化細胞から」と「データ解析から」の2種類のサービスをご提供しています。cDNA合成キット及びライブラリ調製キットについては、東洋紡株式会社で販売中です(詳細>RamDA-seq® kit, Shin-RamDA-seq® kit)。実験デザインの策定や細胞調製等に関するご相談については、弊社でサポートしています。目的とする解析結果を取得するためには、当初からデータ解析を考慮した実験デザインの策定が非常に重要になりますので、ウェット実験を始められる前にまずはご相談下さい。
- 検体やデータはどのように提出したら良いですか?
- シングルセル化細胞をご提出の場合は、専用の細胞溶解液に溶解後凍結し、ドライアイスを同梱し冷凍便にてお送り下さい。発送日は事前にご相談下さい。シーケンスデータの場合は、専用サイトにアップロード頂くか、HDDに保存してお送り下さい。
お問い合わせ
こちらよりお問い合わせください。
専門技術者が原則24時間以内にご連絡します。(土日祝日は除く)
参考文献
Hayashi, T. et al. "Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs", Nature Communications 9(1):619 (2018). https://doi.org/10.1038/s41467-018-02866-0
Ozaki, H. et al. “Millefy: visualizing cell-to-cell heterogeneity in read coverage of single-cell RNA sequencing datasets.”, BMC Genomics 21, 177 (2020). https://doi.org/10.1186/s12864-020-6542-z
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE125288
Matsumoto, M. et al. "Role of PRC2 in the stochastic expression of Aire target genes and development of mimetic cells in the thymus." Journal of Experimental Medicine 222, e20240817 (2025). https://doi.org/10.1084/jem.20240817
*1 RamDA-seq® / Shin-RamDA-seq®は理化学研究所の登録商標です。
*2 参考文献(Hayashi, T. et al.)を基に作成しました。
*3 別途ご相談ください。データにより解析できない場合があります。
*4 Millefyは、理化学研究所と筑波大学の共同研究により開発されたシングルセルのリードカバレッジ可視化用ソフトウェアです。